Kevin W. Paulisse, Tyson O. Friday, Margaret L. Graske, and William F. Polik
Hope College, Holland, MI 49423
Acrolein is a prototypical conjugated bond system which absorbs in the near ultraviolet.
Since acrolein does not fluoresce, Cavity Ringdown (CRD) spectroscopy is used to measure the absorption spectrum of acrolein in a molecular beam.
The S1 vibronic spectrum consists of transitions from vibrational levels of the ground electronic state (S0) to vibrational levels of the first excited singlet electronic state (S1).
The spectrum is assigned by predicting the fundamental transitions using computational chemistry software, analyzing Franck-Condon progressions, comparing spectra taken at different temperatures, and observing rotational band structure.
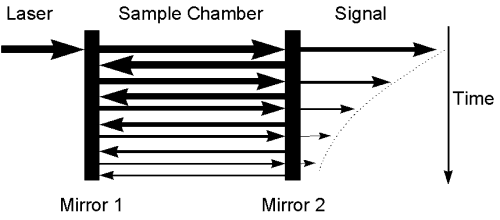
A pulse of laser light is directed into an optical cavity which has highly reflective mirrors on each end. The light bounces back and forth through the sample thousands of times. The small intensity of light that escapes through the second mirror is monitored by a photomultiplier tube (PMT). The ringdown time of the cavity is a measure of absorbance by sample contained between the end mirrors. The large optical pathlength results in a highly sensitive absorption technique.
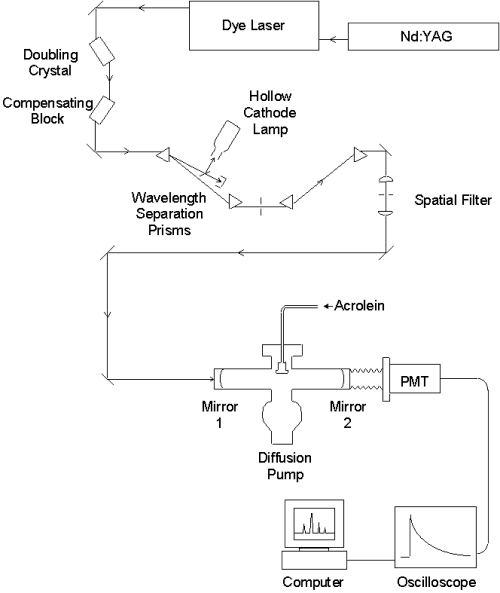
A Nd:YAG laser emits 532 nm light pumping a dye laser, which is tuned over a wavelength range of 680-780nm. The red light passes through a doubling crystal which converts it into ultraviolet (UV) light. The beam translation introduced by the angle-tuned doubling crystal is counteracted with a quartz compensating block. The mixture of UV and residual red light is separated by a pair of 60° prisms, and a second pair of prisms compensates for the beam translation. Some of the red light is directed into a hollow cathode lamp for calibration. The pure UV is directed through a spatial filter, which consists of a focusing lens, pinhole, and collimating lens. The high quality light is sent to the molecular beam chamber where it interacts with the sample. The signal detected by the PMT is averaged by the oscilloscope and sent to the computer, where the lifetime is fit to an exponential decay to monitor absorbance.
The Gaussian 94 program1 was used to predict the frequencies of the S1 normal modes.
Gaussian 94 was first used to compute the S1 normal mode frequencies of formaldehyde (H2CO). Because the systems are similar, frequency scaling factors were calculated by comparing the H2CO results to experimental values from the literature.
Gaussian 94 was then used to compute the S1 vibrational spectrum of acrolein using the RCIS method with the 6-311+G(3DF,2PD) basis set.
| Mode | Description |
Calculated Frequency (cm-1) |
Observed Frequency (cm-1) |
| n1 | Asym vinyl CH stretch |
3097 |
|
| n2 | Symm vinyl CH2 stretch |
3017 |
|
| n3 | Vinyl CH stretch |
3048 |
|
| n4 | Formyl CH stretch |
2866 |
|
| n5 | C=O stretch |
1177 |
1262 |
| n6 | C=C stretch |
1498 |
1406 |
| n7 | CH2 bend |
1316 |
|
| n8 | Formyl CH rock |
1180 |
1132 |
| n9 | Vinyl CH rock |
1316 |
1279 |
| n10 | Vinyl CH2 rock |
867 |
647 |
| n11 | C-C stretch |
1050 |
|
| n12 | CCO bend |
468 |
483 |
| n13 | CCC bend |
301 |
302 |
| n14 | Vinyl C=C torsion |
613 |
|
| n15 | Formyl CH wag |
393 |
|
| n16 | Vinyl CH2 wag |
889 |
916 |
| n17 | Vinyl CH wag |
985 |
|
| n18 | Skeletal torsion |
148 |
252 |
1
Frisch, M., et al. Gaussian 94, Rev. E.2; Gaussian, Inc.: Pittsburgh, PA, 1995.Franck-Condon progressions are series of peaks resulting from changes of a vibrational quantum number in a normal mode. The n18 mode shows a number of Franck-Condon progressions in the room temperature spectrum.
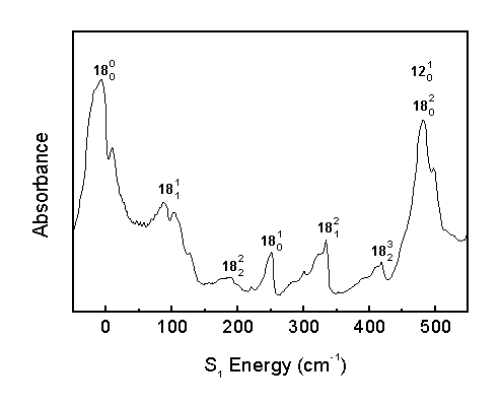
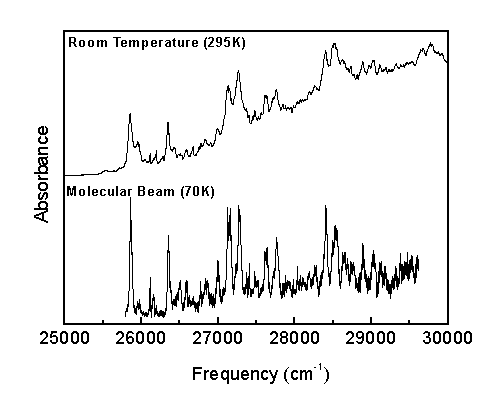
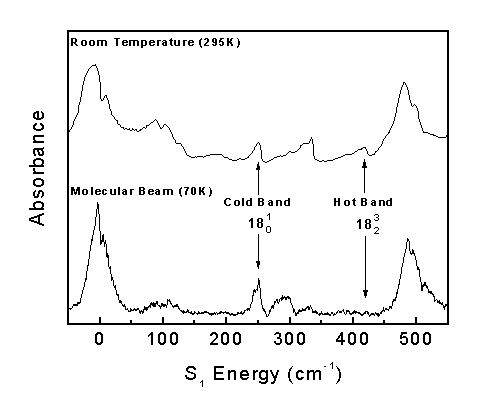
The Boltzmann factor predicts that only the low energy vibrational levels are populated at low temperatures. Consequently, if a band appears in the room temperature spectrum (295 K) but disappears in the molecular beam spectrum (70K), the corresponding transition must have originated from an excited vibrational energy level.
Transitions originating from the ground vibrational level are called "Cold Bands," while transitions originating from excited vibrational levels are called "Hot Bands."
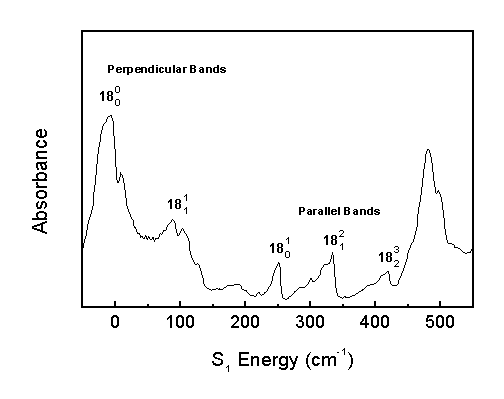
"Parallel Bands" occur when the change in out-of-plane vibrational quanta (n14-n18) is odd. "Perpendicular Bands" occur when the change in out-of-plane vibrational quanta is even.
Parallel bands appear in the spectrum as narrow bands with a sharp high-energy edge, and perpendicular bands appear as broader bands. The narrow parallel bands arise from the D K=0 rotational selection rule (Q-branch), and the broad perpendicular bands arise from the D K=± 1 selection rule (P,R-branches).
The following are the assignments for the acrolein S1 vibronic spectrum. Calculated combination band frequencies are based on an anharmonic oscillator model.
Indicated are the molecular beam S1 energy (MB), room temperature S1 energy (RT), rotational band symmetry (Sym), calculated position (Calc), difference between calculated and observed position (Diff), and the present assignment. The symmetry labels suggested by Brand and Williamson2 are used (a=parallel band, c=perpendicular band).
|
MB (cm-1) |
RT (cm-1) |
Sym |
Calc (cm-1) |
Diff (cm-1) |
Assignment |
|
-573 |
c |
-564 |
-9 |
12(0,1) |
|
|
-328 |
a |
-313 |
-15 |
12(0,1) 18(1,0) |
|
|
-154 |
a |
-160 |
6 |
18(0,1) |
|
|
0 |
0 |
c |
0 |
0 |
Origin |
|
94 |
c |
91 |
3 |
18(1,1) |
|
|
190 |
c |
188 |
2 |
18(2,2) |
|
|
252 |
250 |
a |
251 |
0 |
18(1,0) |
|
297 |
300 |
c |
297 |
0 |
13(1,0) |
|
334 |
334 |
a |
331 |
3 |
18(2,1) |
|
416 |
a |
416 |
0 |
18(3,2) |
|
|
487 |
481 |
c |
487 |
0 |
12(1,0) |
|
487 |
481 |
c |
491 |
-4 |
18(2,0) |
|
514 |
c |
514 |
0 |
X(1,0) |
|
|
564 |
c |
576 |
-12 |
12(1,0) 18(1,1) |
|
|
647 |
648 |
c |
647 |
0 |
10(1,0) |
|
719 |
720 |
a |
719 |
0 |
18(3,0) |
|
732 |
733 |
a |
736 |
-4 |
12(1,0) 18(1,0) |
|
764 |
764 |
a |
765 |
-1 |
X(1,0) 18(1,0) |
|
810 |
a |
814 |
-4 |
12(1,0) 18(2,1) |
|
|
888 |
a |
896 |
-8 |
12(1,0) 18(3,2) |
|
|
911 |
912 |
a |
911 |
0 |
16(1,0) |
|
974 |
965 |
c |
974 |
0 |
12(2,0) |
|
995 |
c |
1001 |
-6 |
X(1,0) 12(1,0) |
|
|
1135 |
1126 |
c |
1135 |
0 |
8(1,0) |
|
1204 |
a |
1199 |
5 |
12(1,0) 18(3,0) |
|
|
1263 |
1262 |
c |
1263 |
0 |
5(1,0) |
|
1294 |
c |
1294 |
0 |
9(1,0) |
|
|
1370 |
a |
1370 |
0 |
8(1,0) 18(1,0) |
|
|
1408 |
1404 |
c |
1408 |
0 |
6(1,0) |
|
1512 |
a |
1514 |
-2 |
5(1,0) 18(1,0) |
|
|
1545 |
1543 |
a |
1545 |
0 |
9(1,0) 18(1,0) |
|
1593 |
a |
1594 |
-1 |
5(1,0) 18(2,1) |
|
|
1616 |
1615 |
c |
1622 |
-6 |
8(1,0) 12(1,0) |
|
1662 |
1664 |
a |
1659 |
3 |
6(1,0) 18(1,0) |
|
1697 |
1700 |
c |
1705 |
-9 |
6(1,0) 13(1,0) |
|
1746 |
1746 |
c |
1746 |
0 |
5(1,0) 12(1,0) |
|
1775 |
1761 |
c |
1776 |
-1 |
9(1,0) 12(1,0) |
|
1896 |
1895 |
c |
1896 |
0 |
6(1,0) 12(1,0) |
|
1985 |
1984 |
a |
1982 |
3 |
5(1,0) 18(3,0) |
|
2017 |
2017 |
a |
2013 |
4 |
9(1,0) 18(3,0) |
|
2063 |
2057 |
c |
2055 |
8 |
6(1,0) 10(1,0) |
|
2120 |
2120 |
a |
2127 |
-7 |
6(1,0) 18(3,0) |
|
2174 |
2175 |
a |
2174 |
0 |
5(1,0) 16(1,0) |
|
2209 |
2208 |
a |
2205 |
4 |
9(1,0) 16(1,0) |
|
2395 |
2395 |
c |
2398 |
-3 |
5(1,0) 8(1,0) |
|
2419 |
c |
2429 |
-10 |
8(1,0) 9(1,0) |
|
|
2544 |
2540 |
c |
2544 |
0 |
5(1,0) 9(1,0) |
|
2544 |
2540 |
c |
2543 |
1 |
6(1,0) 8(1,0) |
|
2632 |
2629 |
a |
2633 |
-1 |
5(1,0) 8(1,0) 18(1,0) |
|
2662 |
2656 |
a |
2657 |
5 |
5(1,0) 12(1,0) 16(1,0) |
|
2669 |
2661 |
c |
2669 |
0 |
5(1,0) 6(1,0) |
|
2685 |
2684 |
c |
2689 |
-4 |
6(1,0) 9(1,0) |
|
2793 |
a |
2795 |
-2 |
5(1,0) 9(1,0) 18(1,0) |
|
|
2872 |
2873 |
c |
2881 |
-9 |
5(1,0) 8(1,0) 12(1,0) |
|
2901 |
c |
2911 |
-10 |
8(1,0) 9(1,0) 12(1,0) |
|
|
2921 |
a |
2920 |
1 |
5(1,0) 6(1,0) 18(1,0) |
|
|
2948 |
a |
2940 |
8 |
6(1,0) 9(1,0) 18(1,0) |
|
|
3022 |
3020 |
c |
3021 |
1 |
5(1,0) 9(1,0) 12(1,0) |
|
3148 |
3156 |
c |
3152 |
-4 |
5(1,0) 6(1,0) 12(1,0) |
|
3168 |
c |
3172 |
-4 |
6(1,0) 9(1,0) 12(1,0) |
|
|
3456 |
3459 |
a |
3455 |
1 |
5(1,0) 9(1,0) 16(1,0) |
|
3603 |
3604 |
a |
3600 |
3 |
6(1,0) 9(1,0) 16(1,0) |
|
3672 |
c |
3679 |
-7 |
5(1,0) 8(1,0) 9(1,0) |
|
|
3811 |
c |
3804 |
7 |
5(1,0) 6(1,0) 8(1,0) |
|
|
3928 |
c |
3936 |
-8 |
5(1,0) 6(1,0) 9(1,0) |
Transition frequencies were calculated using the anharmonic oscillator model with the following fit parameters.
| Parameter | S0 (cm-1) | S1 (cm-1) |
| w5 | 1263 | |
| w6 | 1405 | |
| w8 | 1135 | |
| w9 | 1294 | |
| w10 | 647 | |
| w12 | 564 | 487 |
| w13 | 297 | |
| w16 | 911 | |
| w18 | 168 | 257 |
| wX | 514 | |
| x5,6 | -2.5 | |
| x5,9 | -13.0 | |
| x5,12 | -4.5 | |
| x6,9 | -13.0 | |
| x6,12 | 1.0 | |
| x8,18 | -16.0 | |
| x12,18 | -2.1 | |
| x18,18 | -8.3 | -5.8 |
The observed bands in the room temperature and molecular beam spectra are assigned as shown.
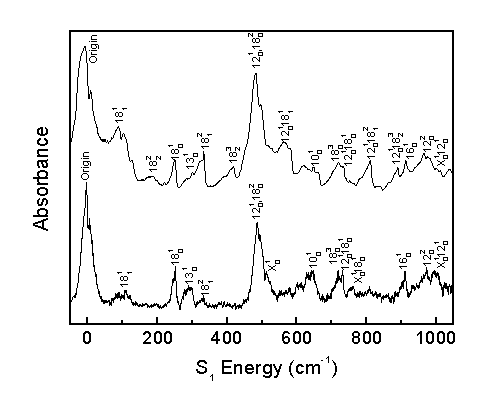
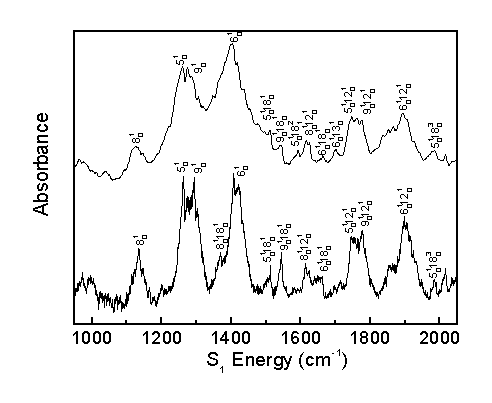
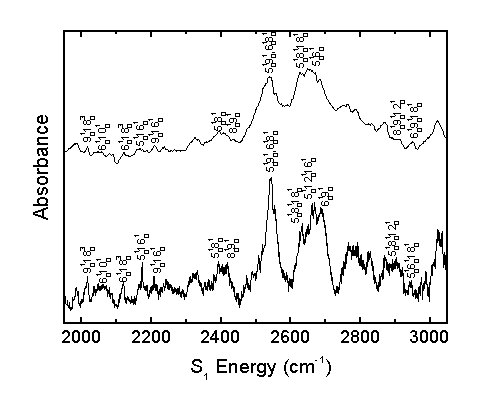
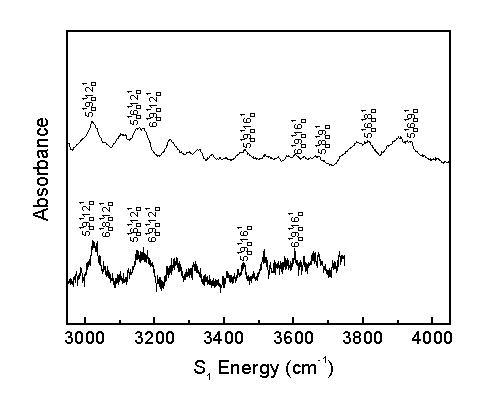
The acrolein S1 vibronic spectrum was assigned considering the computed spectrum, Franck-Condon progressions, temperature effects, and rotational band structure. This allows for the correction of previous literature assignments2 and the identification of previously unassigned bands.
The band previously assigned as 14(1,0) is actually a hot band assigned here as 18(2,1). Consequently, many more combination bands involving n18 are observed than previously believed, allowing for a better understanding of the S1 n18 potential energy surface. The band previously identified as 16(1,0) is also a hot band, but is not yet identified.
The band previously assigned as 15(1,0) is actually a perpendicular band, not a parallel band. This band is assigned here as 10(1,0) based on its computed frequency and rotational band structure.
The band previously assigned as 15(2,0) is assigned here as 9(1,0), as observed progressions indicate two strong fundamental bands in the 1260-1300 cm-1 range.
The present work assigns 65 bands, whereas previous literature2 identified 27 bands, 16 of which were corrected in the present assignment. The present work identifies 9 fundamental transitions in the acrolein spectrum. The identification of numerous other combination bands increases confidence in the assignment of fundamental transitions.
The molecular beam spectrum was critical to correctly assigning the S1 vibronic spectrum by differentiating between hot and cold bands and by simplifying the spectrum to identify bands not observed in the room temperature spectrum.
The computation of the acrolein spectrum provided reasonable predictions of the locations of fundamental transitions. The acrolein results can be used to compute scaling factors for other similar conjugated systems in the future.
We gratefully acknowledge Dr. Marsha Lester (University of Pennsylvania) and Dr. Kevin Lehmann (Princeton University) for useful advice on Cavity Ringdown spectroscopy. We also thank Brad Mulder for fabrication of the apparatus. This research has been sponsored by National Science Foundation grant CHE-9157713 and a research fellowship from Pfizer, Inc.