Assignment
CompChem6: Applications II
1. Bond Orders of Allyl Radical
Build the planar allyl radical C3H5 and perform a Geometry Optimization at the Hartree-Fock 3‑21G level. Note that allyl has an odd number of electrons and is therefore a doublet.
View the output and run a New Job Using This Geometry. Run a Gaussian Bond Order calculation at the same level of theory, which performs a NBO (Natural Bonding Orbital) analysis of the Bond Order. Starting with the same same geometry, run a Gamess Molecular Energy calculation UHF (Unrestricted Hartree-Fock) 3‑21G of this doublet molecule. Finally, starting with the same geometry, run a Mopac Geometry Optimization job at the PM3 level of theory.
If time permits, run a Gaussian Calculation = “AIM=BondOrders” job at the same level of theory. Be patient since AIM jobs take a relatively long time! View the Raw Output to obtain the AIM bond orders.
Construct a table with columns for NBO, GAMESS, MOPAC, and AIM, and rows for each unique bond order and a row for calculation time. Comment on your table.
2. Isodesmic Reaction Analysis of CO2 Heat of Formation
Build and perform Optimize + Vib Freq calculations on carbon dioxide (CO2), formaldehyde (H2CO), and methane (CH4) at the HF/6-31G(d) level. Tabulate the energy (0 K) and enthalpy (298 K) for each.
Use these results to calculate DrxnH for
CO2 + CH4 ® 2 H2CO
Explain why this reaction is an isodesmic reaction. Explain what kind of computational results can be expected for such reactions.
Combine your DrxnH result with the experimental heats of formation for methane and formaldehyde to predict the heat of formation for carbon dioxide. Compare your predicted heat of formation with the experimental value. Experimental heats of formation may be obtained from the NIST Chemistry Webbook (http://webbook.nist.gov/chemistry).
3. Intrinsic Reaction Coordinate Verification of the H2CO ® H2 + CO Transition State
Compute the H2CO ® H2 + CO transition state by building the following planar structure:
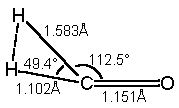
Double check your geometry before proceeding.
Perform a Transition State Optimization of the structure at the Hartree-Fock 3-21G level. View the Results, and then perform a Vibrational Frequencies job with the same geometry and theory. Report the value of any negative (imaginary) frequencies. Note the energy and H‑H bond length.
Using the transition state geometry, perform a Forward IRC calculation at the same theory. View the result and note the energy and H‑H bond length. Use this geometry as the starting point for a Geometry Optimization calculation with the same theory. Note the final energy and the H‑H bond length.
Again using the transition state geometry, perform a Reverse IRC calculation at the same theory. View the result and note the energy and H‑H bond length. Use this geometry as the starting point for a Geometry Optimization calculation with the same theory. Note the final energy and the H‑H bond length.
If either Geometry Optimization calculation job fails, view the Raw Output file and determine why the job failed. Report the line(s) in the output file that indicates the reason for failure. Attempt the Geometry Optimization again, but preview the input file and use “Opt=(MaxCycle=100)” instead of the “Opt” keyword.
Report all numerical values for energy and H‑H bond length in a table. Also, include a sketch of the Potential Energy Surface for the H2CO ® H2 + CO reaction coordinate, along with pictures of the reactant, transition state, and products.
4. Transition State of Diels-Alder Reaction (WebMO Pro)
Addition of cyclopentadiene and acrylonitrile produces the endo norborene product.
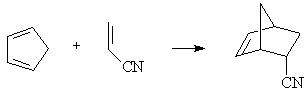
Locate the transition state for this reaction by first building the norbornene product. Perform a PM3 geometry optimization of the product, and verify that the resulting geometry is reasonable.
View the result, choose New Job Using This Geometry, and open the molecule in the WebMO Editor. Adjust the two carbon-carbon bond lengths that are formed in the reaction to 2.2 Angstroms, recalling that the first selected atom is the one that is moved. Open the z-matrix editor with Tools:Z-Matrix.... If necessary change the z-matrix connection definition so that the above two carbon-carbon bonds are directly referenced in the z-matrix (in the Na column) and then click the ReConnect button. Fix both bond length to 2.2 by selecting F (fixed) from the corresponding dropdown menu, and click OK. Perform a PM3 Geometry Optimization calculation on this geometrically constrained molecule.
View the result, and verify that the two carbon-carbon bond lengths formed in the reaction are 2.2 Angstroms. Choose New Job Using This Geometry, and perform a PM3 transition state optimization calculation.
View the result, note the bond length of the two carbon-carbon bonds being formed, and note the near planarity of the cyclopendiene ring. Choose New Job Using This Geometry, and perform a PM3 vibrational frequencies calculation. Verify that there is a single negative (imaginary) frequency that corresponds to the reaction coordinate. Report the value of the negative frequency and an image of the corresponding vibrational mode.
5. Transition States of Diels-Alder Stereoproducts (WebMO Pro)
Substituted reactants undergoing a Diels-Alder reaction can result in various stereoproducts. For example, the addition of acrylonitrile to 5‑fluorocyclopentadiene leads to four possible stereoproducts.
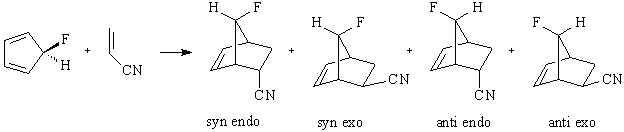
Endo products are generally favored over exo products. Anti products are preferred for alkyl substituted cyclopentadiene, and syn products are preferred for alkoxy or halide substituted cyclopentadiene.
The preferred product can be predicted by comparing the energies of the transition states.
Start with the norbornene transition state in the previous exercise. Open the molecule in the WebMO Editor, and make appropriate substitutions for the transition state to a particular product. Select only the changed atoms, choose Clean-Up:Selection Only, and then choose Clean-Up:Comprehensive. Perform a PM3 Transition State Optimization calculation, followed by a Vibrational Frequencies calculation if desired. Repeat for the other stereoproducts.
Make a table with columns for stereo product and energy. Is the lowest energy transition state consistent with the experimentally observed stereoselectivity?
Optionally, repeat the calculations with 5‑methylcyclopentadiene. Does the lowest energy transition state change?
6. Transition State of Keto-Enol Tautomerization
Acetaldehyde can isomerize to vinyl alcohol via proton migration.
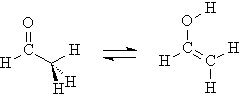
Locate and verify the transition state for this reaction as follows. Build the above conformer of acetaldehyde, and perform a PM3 Geometry Optimization calculation.
View the results, and choose New Job Using This Geometry. Open the molecule in the WebMO editor, and edit the bonds to convert the molecule in vinyl alcohol taking care not to delete or add atoms. Add a bond between the migrating methyl proton to the oxygen atom, adjust the CO double bond to a single bond, adust the CC single bond to a double bond, and delete the bond between the migrating methyl proton and the carbon atom. [It is critical to not delete or add any atoms so that the same atom ordering and z-matrix definition is used for both molecules.] Choose Clean-Up:Hybridization and Clean-Up:Geometry. Perform a PM3 Geometry Optimization calculation of this vinyl alcohol molecule. Note the job number of this vinyl alcohol calculation.
View the results of the acetaldehyde job again, and choose New Job Using This Geometry. Perform a PM3 Saddle Calculation. Before submitting the job, click the Advanced Options button, and enter the job number of optimized vinyl alcohol as the Second Geometry. Close the Advanced Job Options, and submit the job.
View the results of the saddle calculation, choose New Job Using This Geometry. Perform a PM3 Transition State Optimization calculation.
View the results of the trasition state optimization job, and choose New Job Using This Geometry. For cosmetic purposes, open the molecule in the WebMO editor and adjust the bonds in the HOCC 4-membered rings to single bonds. Perform a PM3 Vibrational Frequencies calculation. Verify that there is a single negative (imaginary) frequency, and report its frequency. Verify that the corresponding vibrational mode corresponds to proton migration. What other motions are involved at the transition state?
Optionally, verify that this geometry is a transition state by performing Forward and Reverse IRC calculations, optimizing the resulting geometries, and comparing them to the reactant and product.
7. With over 2,000 licenses granted to date, WebMO version is rapidly gaining popular acceptance in the chemistry community. We are interested in any suggestions you might still have for WebMO, particularly involving its usability, View Results report, and user interface, to make it even more user-friendly and useful.