Assignment
CompChem5: Applications I
1. Partial Charge Analysis of Electrophilic Aromatic Substitution Reactions
The observed isomeric product distribution of electrophilic attack by NO2+ on nitrobenzene and chlorobenzene can be analyzed by examining the partial charges on the otho, meta, and para carbons of substituted benzene reactant.
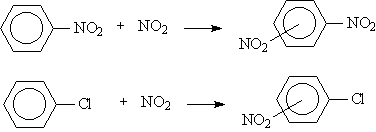
|
Product |
ortho |
meta |
para |
|
C6H4N2O4 |
6% |
93% |
1% |
|
C6H4NO2Cl |
30% |
1% |
69% |
Build nitrobenzene such that the nitro group lies in the molecular plane and has equal NO bond lengths. Pre-optimize this structure with a PM3 calculation. If using Gaussian, disable symmetry using the Advanced Job Options. [The NoSymmetry keyword prevents the job from terminating if the molecular symmetry changes during calculation.] Optimize the geometry of nitrobenzene at the HF/3-21G level, also disabling symmetry for the calculation. The reported partial changes are Mulliken partial charges.
If using Gaussian, use the New Job Using This Geometry button to perform a Single Point calculation at same level of theory with the additional keyword “Pop=NBO”. [The Pop=NBO or Pop=NPA keywords request that Gaussian perform a Natural Bonding Orbital analysis of the molecule, in which electrons are assigned to “organic chemistry” orbitals as core electron pairs, single bonds, double bonds, lone pairs, etc.] Finally, use the New Job Using This Geometry button to perform a Single Point calculation at same level of theory with the additional keyword “Pop=CHelpG”. [The CHelpG keyword requests that Gaussian calculate an electrostatic potential-derived potential in which atoms are assigned partial charges to match the electrostatic potential at the van derWaals surface.] View the Raw Output of each of these jobs and locate the respective partial charges.
Construct a table with columns for isomer (ortho, meta, para), Mulliken partial charge, NBO partial charge, ChelpG partial charge, and observed nitration product distribution percentage. Use these results to explain the preferred site of electrophilic attack by NO2+.
Repeat the above calculations and construct a similar table for chlorobenzene.
Intuitively explain the computed partial charges by drawing resonance structures for nitrobenzene that withdraw p-electron density from the ring and by drawing resonance structures for chlorobenzene that donate p-electron density into the ring.
Comment on the differences among the methods for computing partial charge.
2. Carbocation Intermediate Analysis of Electrophilic Aromatic Substitution Reactions
Aromatic substitution reactions proceed via a carbocation intermediate. The product distribution of an aromatic substitution reaction may be determined by the relative stabilities of the carbocation intermediates. The aldehyde group is meta-directing for aromatic substitution reactions.
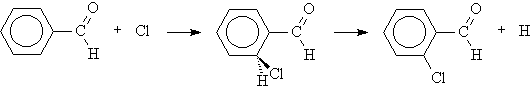
|
Product |
ortho |
meta |
para |
|
C6H4CHOCl |
19% |
72% |
9% |
Build planar benzaldehyde and optimize the geometry with a PM3 calculation. View the partial charges of the ortho, meta, and para carbons (on the side of the ring closest to the H atom of the aldehyde group).
Click New Job Using This Geometry, and open the molecule in the WebMO editor. Add a chlorine atom to the ortho carbon (on the H atom side of the aldehyde group). Adjust the double bond to the ortho carbon to a single bond. Select the CHCl group, choose Clean-Up: Selection Only, and choose Clean-Up: Comprehensive so that only the new change is cleaned up. Optimize the geometry of this intermediate structure with a PM3 calculation, setting the charge appropriately for a carbocation. Repeat this to compute the PM3 energies of the meta and para intermediates.
Construct a table with columns for isomer (ortho, meta, para), partial charge, intermediate energy, and observed chlorination percentage.
Comment on your results.
3. Rigid Potential Energy Scan of CH Bond Dissociation
Build CH4 and clean up only its geometry. Use Tools: Z-Matrix and Fix All coordinates. C1-H5 bond length from 0.7 to 2.7 in 20 steps. Perform a Coordinate Scan calculation at the HF/STO3-G level.
Repeat the coordinate scan with various levels of theory and basis sets, up to MP2/6‑311+G(p,d).
Download the coordinate scans and use Excel to plot these potential functions in the same figure. Comment generally on the differences among these plots.
Calculate the CH bond dissociation energy in kcal/mol by taking the difference between the minimum and separated energies. Compare your results to an experimental value from a general or organic chemistry textbook.
4. Relaxed Potential Energy Scans of NH3 and H2O Inversion
Build NH3 with the WebMO editor. Use Tools: Z-Matrix to scan the H4-N1-H2-H3 dihedral angle from 90 to 270 in 18 steps. Perform a Coordinate Scan calculation at the PM3 level. From the results page, view the Coordinate Scan and determine the barrier height for ammonia inversion in kcal/mol.
Repeat the calculation for H2O, scanning the H3-O1-H2 bond angle from 90 to 270 in 18 steps. View the Coordinate Scan and determine the barrier for water linearization in kcal/mol.
Speculate on the reason for difference in barrier heights.
Repeat one the above calculations as a rigid scan, holding all variables fixed except for the scanned variable. How does the barrier height compare to the previously computed barrier height? Explain the reason for the difference.
5. How could WebMO be improved to assist you with your calculations? Please be as imaginative and thorough as possible with your suggestions and constructive criticism!!