Assignment
CompChem3: Frequency Calculations
1. Normal Vibrational Modes of Formaldehyde.
Build formaldehyde, H2CO, and perform a Geometry Optimization PM3 calculation on it. View the result, click on New Job Using This Geometry, and perform a Vibrational Frequencies PM3 calculation. View the result, and view each vibrational mode. Fill in the following table of vibrational frequencies by visually identifying the description of each normal mode
|
|
|
PM3 |
Literature |
|
n1 |
sym CH stretch |
|
2811 |
|
n2 |
CO stretch |
|
1756 |
|
n3 |
CH2 bend |
|
1500 |
|
n4 |
out-of-plane bend |
|
1170 |
|
n5 |
antisym CH stretch |
|
2861 |
|
n6 |
CH2 rock |
|
1251 |
2. Stationary Points of Vinyl Amine.
Build planar and pyramidal vinyl amine, CH2=CHNH2.
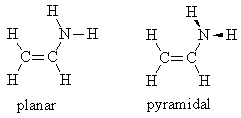
For planar vinyl amine, adjust the hybridization of N to sp2 before cleaning up the geometry. Be sure to adjust the dihedral of the amine group appropriately for each molecule. For each conformation, optimize the geometry and calculate vibrational frequencies with a PM3 (or Hartree-Fock 6‑31G(d) or better) calculation.
Make a table with columns for conformation, energy, and type of stationary point. Characterize each stationary point as a minimum or transition state using the frequency results.
Include a picture of the vibrational mode corresponding to the reaction coordinate for inversion about the nitrogen atom.
3. Stretching Frequencies of Carbonyl Groups.
Calculate the C=O stretching frequency of formamide, acetaldehyde and acetyl chloride.
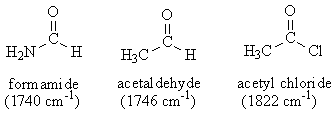
Perform Optimize Geometry and Vibrational Frequencies (or Optimize+Vib Freq) Hartree-Fock 3‑21G calculations for each molecule. Computed vibrational frequencies are systematically high and are routinely scaled need to be scaled frequencies. The scaling factor for HF/3-21G calculations is 0.9085. Make a table with columns for molecule, calculated CO stretch frequency, scaled CO stretch frequency, and experimental frequency (indicated above).
Comment on the absolute and relative accuracy of frequency calculations.
4. Transition State for 1,3 Hydrogen Shift of Fluoropropene.
Build the transition state for the 1,3 hydrogen shift of fluoropropene.
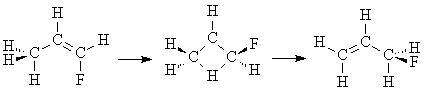
Do not do a comprehensive cleanup, as the Add Hydrogens function uses rules that are applicable to stable molecules, not transition states. Instead, manually add the hydrogens, including a hydrogen that is bonded to both C1 and C3. Adjust the hybridization of C2 to sp2, and clean up the geometry only. Adjust the H-C2 bond angle appropriately so it is symmetric with respect to the ring. Perform an Optimize Transition State PM3 (or Hartree-Fock 3-21G) calculation on the molecule. If the job was successful, view the result to see if the molecule looks as expected. If the job failed, rebuild the transition state but try adjusting the C-C-C bond angle to approximately 100° while maintaining the symmetry of the ring (i.e., increase C1-C2-C3 by 15°, and then increase C3-C2-C1 by 15°).
Create a new job using the optimized transition state geometry. Perform a Frequency PM3 calculation on the previously optimized geometry. View the transition state.
Insert a picture of the transition state and reaction coordinate. How do you know that it is a transition state?
Build and optimize the geometries of the reactant and product for this reaction. Make a table with columns for species and energy, and include the energies of the reactant, transition state, and product. What is the reaction barrier for 1,3 hydrogen shift of fluoropropene?
5. Thermochemistry of CH3CHO ® CO + CH4 in the gas phase.
Build and perform Optimize+Vib Freq Hartree-Fock, 6‑31G(d) calculations on acetaldehyde, carbon monoxide, and methane. To speed up your calculation, start your acetaldeyhde calculation from the same geometry as a previous lower level optimization.
Make a table of calculated values with columns for molecule, Cv, S, E298, H298, and G298.
Visit the NIST webbook (http://webbook.nist.gov/chemistry) and make a table of experimental values with columns for molecule, Cp, S, and DfH (kcal/mol).
Compute Cp from your calculations for each species by using the relationship that Cp = Cv + R for an ideal gas.
Compute DrxnH from your calculations by appropriately combining H298 values and converting to kcal/mol. 1 Hartree = 627.5095 kcal/mol. Compute DrxnH from the experimental data by appropriately combining DfH.
Comment on the agreement between calculation and experiment by comparing Cp, S, and DrxnH values in a table.
6. How could WebMO be improved to assist you with your calculations? Please be as imaginative and thorough as possible with your suggestions and constructive criticism!