Assignment
CompChem2: Geometry Optimizations
1. Conformers of n-Butane.
Use the WebMO Editor to build n‑Butane. Set the initial C1-C2-C3-C4 dihedral angle to 30°. Perform a geometry optimization calculation on the molecule, and record the optimized dihedral angle and energy. Repeat the calculation with an initial dihedral angle of 150°.
Explain why different structures result from the two optimizations. Characterize each optimized structures as either a local or global minimum.
2. Potential Energy Surface of Methyl Rotation in n-Butane (WebMO Pro).
Build and clean-up n‑butane with an initial dihedral angle of 180°. Open the Z-matrix editor with Tools: Z-Matrix.... First, verify that the four carbons are listed first and that a terminal carbon atom is the fourth atom (for which a dihedral angle is defined). If this is not the case, reorder the atoms appropriately in Order column and click the ReOrder button. Second, change the Dihedral Opt entry for the terminal carbon from O (= optimize) to S (= scan). Third, define the scan by setting Start to 0, Stop to 180, and # Steps to 18. This will result in 19 calculations as the dihedral angle takes on values of 0, 10, 20, ..., 180. Click OK to close the Z-Matrix editor.
Perform a Coordinate Scan calculation with PM3 theory. If using MOPAC, click the Advanced Options button and specify the additional keyword PRECISE. Preview the input file and note how the scanned coordinate is specified.
After the job is completed, view the results and view the coordinate scan. Indicate where the trans and gauche conformers on the potential energy surface plot. Explain the difference in the energies of these two conformers.
3. VSEPR Theory.
Calculate the energies of planar, tee, and pyramidal ClF3 by performing geometry optimization calculations using Hartree-Fock theory with the STO-3G basis.
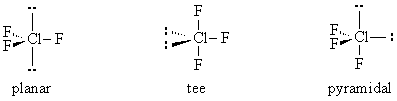
To build a particular geometry, construct ClF5, set the hybridization of the central Cl atom to dsp3 with the Adjust tool by control-clicking on Cl, choose Clean-Up: Geometry (not Comprehensive!), and finally delete the undesired F atoms.
If a particular geometry optimization calculation fails to converge, click the Restart button in Job Manager, and view the last computed geometry. If this is different than the initial geometry, what does this mean about the stability of the initial geometry?
Construct a table with columns for initial geometry and optimized energy. Explain whether or not your results agree with VSEPR (Valence Shell Electron Pair Repulsion) theory learned in General Chemistry.
4. Conformers of Vinyl Alcohol.
Build vinyl alcohol-0°, vinyl alcohol-180°, acetaldehyde-0°, and acetaldehyde-60°.
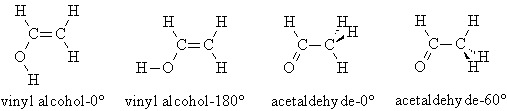
Perform a Geometry Optimization, Hartree-Fock 3-21G calculation on each. Make a table with columns for molecule, number of optimization steps, energy (Hartree), relative energy (kcal/mol). The relative energy should be calculated relative to the global minimum. Note that 1 Hartree = 627.5095 kcal/mol. For one of these jobs, report the keywords that control the type of calculation being run. Also report the text from the corresponding output file indicating that the optimization has converged.
5. Transition State of Vinyl Alcohol.
Build an approximate transition state for the isomerization of vinyl alcohol-0° and vinyl alcohol-180° as follows. View a geometry optimized vinyl alcohol job that successfully ran, and choose New Job Using This Geometry. Adjust the H-O-C=C dihedral angle to 90°. Perform a Transition State Optimization, Hartree-Fock 3-21G calculation.
What are the keywords in the input file for this calculation? What indications are present in the output file to suggest that this optimization differs from the previous optimizations.
Make a table with columns for conformation and relative energy (kcal/mol). Include the two stable conformations and the transition state in the table.
6. Transition State of Silane Elimination Reaction.
In this exercise, the transition state for the reaction SiH4 ® SiH2 + H2 will be found.
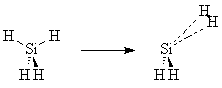
Build SiH4 and perform either a PM3 or a Hartree-Fock 3-21G Geometry Optimization on it. After the calculation has completed, note its job number, and view the job. Choose New Job Using This Geometry. Adjust the geometry of the molecule as follows. Adjust the bond length two Si‑H bonds to 3.0 Angstroms. Adjust the H‑Si‑H bond angle for these two lengthened bonds to 15°. Verify that the distance between these two H atoms is now 0.78, which is similar to the H2 bond length of 0.74 Angstroms. Perform a Saddle Calculation at the level of theory used for the previous geometry optimization. Before submitting the job, click the Advanced Options button, and enter the job number of the previous optimization for the Second Geometry. If using MOPAC, specify the additional keyword GEO-OK (which allows the calculation to proceed despite the short distance between the H atoms). Close the Advanced Job Options window. Check Preview Input File and submit the job.
Describe the general appearance of the input file. Specifically, how does it differ from other input files? Submit the job.
Provide a picture of the transition state for this reaction. Which H atoms are forming molecular hydrogen? What is their bond length, and how does that compare to the atom distance between the two H atoms in SiH2? How does their bond angle with Si compare to the SiH2 bond angle?
Describe how the transition state search algorithm for this exercise differs from the previous exercise. When would you use each algorithm?
7. How could WebMO be improved to assist you with your calculations? Please be as imaginative and thorough as possible with your suggestions and constructive criticism!