Assignment
CompChem1: WebMO and Energy
Calculations
1. Creating structures using WebMO.
Use WebMO Editor to build the following molecules.
|
A. Propene, CH3CH=CH2 |
|
|
B. Aspirin, C9H8O4 |
|
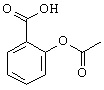
Use the Fragment Library (Build: Fragment...) in the WebMO Editor to create the following molecules. When working with two fragments, it is often useful to select one fragment, and then rotate or translate just the selected fragment with Adjust: Rotate Selection or Adjust: Translate Selection. It is also useful to cleanup just a portion of the molecule by selecting the atoms for cleanup, choosing Clean-Up: Selection Only and the choosing Clean-Up Comprehensive.
|
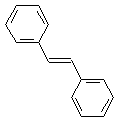
C. Trans-stilbene, C6H5CH=CHC6H5 |
|
|
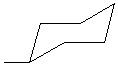
D. Equatorial methylcyclohexane (chair conformation), CH3C6H11 |
|
From the WebMO Build Molecule page, import the following molecules as chime structures. Locate these structures by performing an internet search for the terms “chime” and “molecule”, where “molecule” is the molecule of interest. When viewing these molecules in a web browser as chime structures, save them to you local hard disk with right-click: File: Save Molecule as... locally in either MOL or PDB format. On the WebMO Build Molecule page, click the Import Molecule button, choose the proper format, and browse to the saved file.)
E. Taxol, C47H51NO14
F. Buckminsterfullerene, C60
Useful Tip: In Windows, Alt-Print Scrn copies the active window to the clipboard as a graphic bitmap image. This is very useful for inserting WebMO images into other applications, such as Microsoft Word or PowerPoint. It is often preferable to change the WebMO Editor background color (File: Preferences...) from gray to white. Also, the resulting image usually needs to be cropped. This can be done within Word or PowerPoint with the crop tool on the Picture Toolbar. Or it can be done by pasting the image into an image-editing program, such as the Paint program in Windows. Activate Paint, choose Edit: Paste to paste the image into Paint, use the rectangle tool to select the region surrounding the molecule, choose Edit: Copy... to copy the rectangle to the clipboard (or Edit: Copy to... to save the rectangle to disk), and finally activate Word and paste the rectangle into Word.
2. Previewing input file.
Build formaldehyde, H2CO, using WebMO. Perform a single point Hartree-Fock 3-21G calculation on it. Check the Preview Input File checkbox before submitting the job. Identify the purpose of each part of the input file.
3. Locating results in raw output file.
Build formaldehyde, H2CO, using WebMO. Perform a single point Hartree-Fock 3-21G calculation on it. View the results using WebMO. Also view the raw output file by clicking the Raw Output button. Make a table that lists each item on the WebMO results page and the corresponding line(s) in the raw output file. Make a list of items in the raw output file that are not listed on the WebMO results page.
4. Dipole moment of propene.
Build propene, CH3CH=CH2, using WebMO. Perform a single point Hartree-Fock 3-21G calculation on it. View the dipole moment of propene. Note that WebMO follows the chemistry convention of the dipole moment pointing from positive to negative, i.e., towards excess electron density. Speculate on the source of the dipole moment in propene.
5. Formaldehyde molecular orbitals
Build formaldehyde, H2CO, using WebMO. Perform a molecular orbital Hartree-Fock STO-3G calculation on it. Repeat using the 3-21G basis set.
Characterize the following sets of orbitals:
· the two lowest energy orbitals
· the remaining occupied orbitals
· the unoccupied orbitals
View the HOMO and LUMO, and describe each orbital (sigma, pi, bonding, anti-bonding, etc.).
What atomic orbitals are used for row 2 atoms (O and C) in each basis set? For H atoms?
6. C13 NMR shifts of butane, butene and butyne.
Build butane, butene and butyne. Adjust the dihedral angles to result in planar, staggered conformations. Perform an NMR Hartree-Fock 3-21G calculation on each.
If any of the jobs failed, view the Raw Output from Job Manager and determine why the job failed. Report the line(s) in the output file that indicates the reason for failure, and explain why this happened. Build the molecule again and re-run the job, but on the Configure Job Options page, click the Advanced Options button and choose Cartesian Coordinates.
Gaussian computes absolute chemical shifts. WebMO converts these chemical shifts to shifts relative to TMS. Make a table with columns for compound, C atom (outer or inner), absolute shift, relative shift, and literature value. Justify these chemical shifts based on your chemical knowledge.
|
compound |
Couter |
Cinner |
|
butane |
13.4 |
25.2 |
|
trans-2-butene |
17.6 |
126.0 |
|
2-butyne |
— |
73.6 |
|
Silverstein, Bassler, and Morril, Spectroscopic Identification of Organic Compounds, 5th ed. (Wiley, New York, 1991, Tables 5.2, 5.6, 5.8 |
||
7. Please offer any constructive criticism you may have on how WebMO could be improved to assist you with your calculations.