
![]()
![]()
![]()
![]()
![]()
A brief description of the TDC method along with some background on traditional methods is given below. A more complete description may be found in Krueger, B. P., et al. J. Phys. Chem. B 1998, 102, 5378-5386. (See also the corrected figure in J. Phys. Chem. B 1998, 102, 9603).
The impetus for developing the TDC method was experimental work in the 1990s that was revealing the rapid timescales of electronic energy transfer (EET) between the pigment molecules involved in photosynthesis (for a review see for instance Sundström, V., et al. J. Phys. Chem. B 1999, 103, 2327-2346) To model these results, it is necessary to understand the balance between several factors, one of which is the electronic coupling between the transition moments of the pigments.
The traditional method for calculating this coupling strength was developed by Förster in the late 1940s (Förster, T. Ann. Physik 1948, 2, 55-75). In his treatment, Förster assumed that the total electronic coupling was dominated by the Coulombic coupling and that the Coulombic coupling between molecules could be reasonably represented by the transition dipole-transition dipole coupling
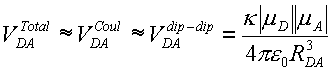
where |µD| and |µA| are the magnitudes of the donor and acceptor transition dipoles, RDA is the distance between them, and the orientation factor is given by
![]()
where the hats denote unit vectors. The first assumption, that the total coupling is well-represented by the Coulombic coupling, results in small errors when the molecules have significant orbital overlap. Note that the TDC method calculates the Coulombic coupling, not the total coupling, so orbital overlap effects must be considered separately (see Scholes, G. D. et al. J. Phys. Chem. B 1999, 103, 2543-2553). The second approximation, the ideal dipole approximation (IDA), causes significant errors in many more cases. The IDA breaks down when RDA is small -- roughly the same size or smaller than the molecules involved -- such that Vdip-dip may not be an accurate representation of VCoul. This breakdown occurs because the molecular transition moments no longer appear point-sized and because the centers of the transition moments, from which RDA is measured, become difficult to define. Molecules with extended (such as carotenoids) or asymmetric transition densities (such as those shown on the TDC home page) exaggerate errors in the IDA, making it inappropriate for photosynthetic systems.
Therefore, we needed to develop a more robust method for estimating the Coulombic coupling. The result is the transition density cube (TDC) method described at this site, in which a quantum mechanical calculaiton is used to determine wavefunctions for the initial and final states of the molecular transitions involved. The inverse product of these wavefunctions is the transition density
![]()
where g and e denote the ground and excited states of molecule k. The Coulombic interaction between the donor and acceptor transition densities gives the exact Coulombic coupling (equivalent to retaining all terms in the multipole expansion).

Computationally, we use the quantum mechanical package to integrate each transition density into an array of finite-sized volume elements
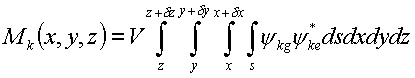
where V is the element volume. The set of finite-sized volume elements for a molecule is called the transition density cube (TDC). To calculate the Coulombic coupling between two TDCs, the Coulombic interactions between all the elements of each cube are summed,
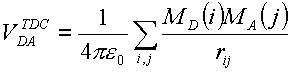
where i and j denote the elements (x,y,z) of the donor and acceptor TDCs and rij is the distance between elements i and j.
The TDC method allows a highly accurate estimate of the Coulombic coupling that takes the three dimensional shape of the transition moments into account and is valid for any intermolecular separation. Because the complete transition density is used, without employing a multipole expansion, there is also no need to arbitrarily specify the centers of the molecules. Accuracy of the method is limited only by the accuracy of the wavefunctions and by the sizes of the volume elements of the TDCs. In most implementations the TDC method represents a dramatic improvement over the IDA and intermediate (e.g. monopole) methods and is computationally tractible for large, biologically relevant molecules such as hemes, chlorophylls, and carotenoids.